EcoSNP-VISTA
See also: GeneSNP-VISTA - Visualization of mutations in genes
System requirements
EcoSNP-VISTA can be executed on any Windows Linux or MacOS X computer with installed Java version 1.4 or higher.
Follow these instructions to install Java on your machine:
Windows |
Linux |
Solaris
Macintosh users should upgrade their OS to 10.1 or better. Additional java upgrades may be available from Apple's download page.
Installation instructions
- download the self-extracting archive for Windows, Linux or MacOS X
- Save the file on your desktop
- For Mac unzip and untar the archive EcoSNP-VISTA.mac.tar.gz which will create EcoSNP-VISTA.mac directory. The program will create a folder EcoSNP-VISTA.mac with several files. After this the archive can be removed.
- For Linux unzip and untar the archive EcoSNP-VISTA.linux.tar.gz which will create EcoSNP-VISTA.linux directory. The program will create a folder EcoSNP-VISTA.linux with several files. After this the archive can be removed.
- For windows start the EcoSNP-VISTA.exe program by clicking on the icon. The program will create a folder EcoSNP-VISTA with several files. After this the archive can be removed..
- Windows user can run program by executing the start.bat file
Linux and Mac Users must execute the start.sh file
in EcoSNP-VISTA.mac directory. For a demo use demo.sh. Note for UNIX users: files cluster and calculateBreakpoints must be executable.
User manual
Input files
| File | status | description |
|---|---|---|
| BLAST | mandatory | BLAST output file is used as an input file for the SNP-VISTA program. Distributive package contains an example file Results.cgi.txt |
| Recombinational points | optional | This text format file contains a list of recombination points. Each line of this file contains read name followed by coordinate of recombination point in query coordinates. See example file for details |
| annotation | optional | This text format file contains annotation. Each line of the file contains description of one annotated region and consists of three fields separates by space: annotation_type start_pos_on_query end_pos_on_query. See example file for details |
The distributive package contains some example files. The start.bat will rum SNP-VISTA with these input files: -f Results.cgi.txt -a annot.txt -b breakpoints.txt and a user can edit the start.bat file by using the right button of the mouse and replace the name of the files by yours. If program will found that some mandatory files were omitted it will open a Wizard window so you can open necessary files at program startup.
See Usage section for details.
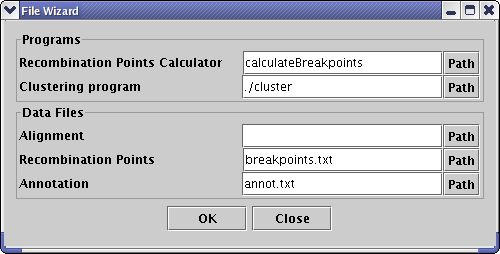
| Relation between wizard and command line parameters | |
|---|---|
| Wizard parameter | Command line parameter |
| Recombination Points Calculator | -calc |
| Clustering program | -cl |
| Alignment | -f |
| Recombination Points | -b |
| Annotation | -a |
Output files
Recombination points files are produced by the program started with the BP button on the top panel of the tool. This is a text format files with a list of break points. Each line of this file contains read name followed by coordinate of recombination point in query coordinates. See example file for details
Overview of the display
Click on any part of the image to see details
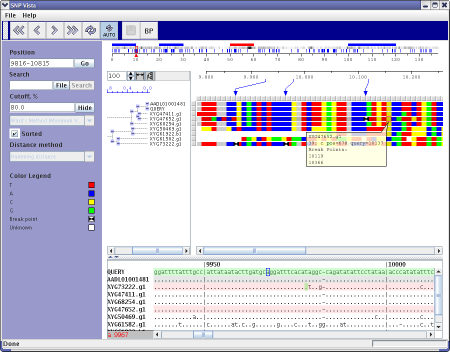
Toolbar

 |
 |
Scroll left or right 1/2 of window width from the current QUERY position |
 |
 |
Scroll to a previous or next position on the QUERY |
 |
Recalculate clustering tree in the tree panel and rebuild the view | |
 |
Enable or Disable automatic tree calculation when position is changed | |
 |
 |
Save Break Points created by user in the current recombination points file or you can select other file name |
| BP | Open Recombination Points Calculator window. |
Control Panel
Control Panel contains the following text fields:
|
Coordinate ruler

The top panel of the program shows annotated regions, SNP density, recombination points relatively to the QUERY sequences coordinates. Small red rectangle shows current position relative the whole query length. You can drag this rectangle by mouse to change current position or drag left or right rectangle borders to change start or end position respectively. Small red triangles show all recombination points for the selected read panel.
Tree Panel
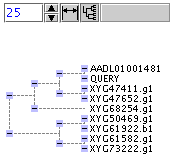
Tree panel reflects results of clustering procedure. Hamming distance between different sequences is calculated as a relative number of matches of two sequences for each position in the interval. Tree panel is mouse clickable. Each click on small blue rectangles will hide corresponding branch of the tree. Repetitive click in the same rectangle will show hidden branch. After each tree recalculation tree panel will show the whole tree with all branches visible.
Control elements of tree panel are used to enable or disable auto-fit mode and absolute/relative distance mode.
 Pressing this button switch between absolute/relative distance modes. In absolute distance mode each branch of the tree has length reflecting real distance between sequences. In relative mode all branches have the same length and tree reflects only relative positions of sequences to each other
Pressing this button switch between absolute/relative distance modes. In absolute distance mode each branch of the tree has length reflecting real distance between sequences. In relative mode all branches have the same length and tree reflects only relative positions of sequences to each other
 If this button is pressed, Tree panel will try to adjust its width to the maximum tree width after all tree recalculation.
If this button is pressed, Tree panel will try to adjust its width to the maximum tree width after all tree recalculation.
Text control contains a scale number. Increasing or decreasing of this number will increase or decrease tree width respectively.
Zoom Panel
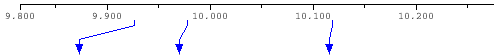
Zoom panel shows positions of recombination points on the zoomed area of the QUERY sequence and points to their positions on the SNP map panel.
SNP Panel
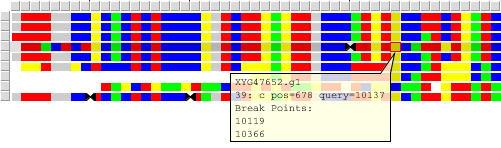
Each missmatch between query sequence and other sequences is shown in the SNP panel as a colored square. Color of the square shows exact letter in this position of the sequence. (See legend panel for details)sand-glass sign points to a recombination point near SNP position.
Mouse click on any square will show in popup window information about this position and recombination points on the sequence.
Data Panel
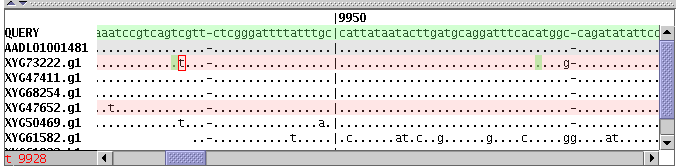
Each line of the Data panel contains one sequence in query coordinates. Order of lines is coincided with order of curves in tree and SNP panels. The following color schema is used:
Light green bar - query line
Pink bar - sequence that contains at least one recombination point in the selected interval.
Gray bar - sequence contains some recombination points, but they are outside the selected region.
When mouse cursor is moved inside the panel and cross gray or pink bars breakpoint will be shown as red triangles in coordinate rule, even outside selected region.
Position highlighted with green rectangle is a recombination point. When mouse cursor moves above sequences letters red frame highlites corresponding position. Position coordinates are shown in the left gray panelnext to horizontal scrollbar
Custom operations with recombination points
|
Status Bar

Status Bar is used to show current status messages and contains progress bar
Recombination Points Calculator
Recombination points calculator is a front-end shell for the breakpointsCalculator program.
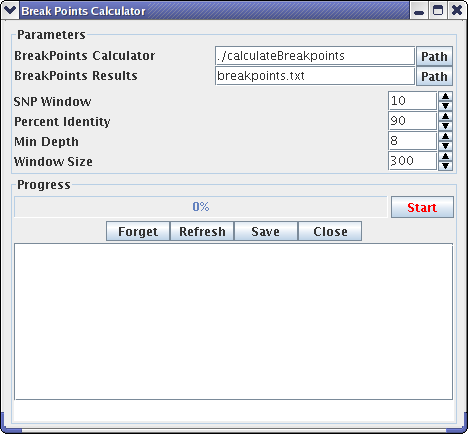
Parameters:
Buttons:
|
Usage
java [-Xmx###M] -classpath "... ..." DTree parameters
For classpath details see corresponding starting script.
| Parameter | Default | Comment |
|---|---|---|
| -cl | ./cluster | Path to cluster program |
| -calc | ./calculateBreakpoints | Path to Break Points Calculator |
| -f | - | BLAST Data File |
| -b | - | BreakPoints File |
| -a | - | Annotation File |
| -cut | 80 | Cut off, % |
| -d | Debug output |
Troubleshooting
- starting scenario assumes that java executable file is available from the standard path
If it's not so you can either add your java directory to your path or you can edit scenario file (start.bat or start.sh) and replace the "java" by its absolute path - Not enough memory error:
You can increase amount of memory available to Java machine by changing the -Xmx###M parameter.
### must be replaced by amount of memory in megabytes. Edit start.sh or start.bat
file to increase this parameters if default value -Xmx350M is not enough for your data set. - UNIX and MacOS X users: Program starts but can nor build cluster tree or can not calculate breakpoints: Probably, file DTree/cluster or DTree/calculateBreakpoints program is not executable.
use the following commands:
cd DTreeRestart the program
chmod u+x ./cluster
chmod u+x ./calculateBreakpoints
Questions/Comments please email to vista@lbl.gov