Using mVISTA web-interface
- Input to the server
- Required Fields
- Optional Fields
- Results
-
Input to the server
At the first page you will be asked to identify the number of genomic sequences you want to analyze. Entering this number and clicking "submit" will take you to the main submission page which will contain the number of fields corresponding to the number of sequences you entered.
The mVISTA server can process up to 100 sequences.
Required Fields
E-mail address
We ask for you email address so that we can notify you when the results are ready.
Sequences
You can submit your sequences to the server two ways:
-
Upload them from your computer as a plain text file in Fasta format using "Browse" button. The DNA sequence of the base organism has to be submitted as one contig (can be finished or ordered/oriented draft merged into one contig) while the other species' sequences can be in one or in several contigs (draft).
Sample sequence in Fasta format (you will find more details on the format at the NCBI site):
>mouse ATCACGCTCTTTGTACACTCCGCCATCTCTCTCT CTCTCGAGCAGATCTCTCTCGGGAATATCGACAA ...
 Note: at this time we accept only the letters CAGTN and X in your sequence. Please make sure to submit a sequence as plain text, not a Word or HTML file.
Note: at this time we accept only the letters CAGTN and X in your sequence. Please make sure to submit a sequence as plain text, not a Word or HTML file.If you submit your sequences in FASTA format, we suggest that you use meaningful names for them since these names will appear on the plots we generate. If you work with a draft sequence, each of the contigs will be named the way you indicated after the ">" symbol.
-
You can specify its GenBank accession number, which will be used to automatically retrieve the sequence from the GenBank database and process on our server.
In both cases, the total size of the sequences should not exceed 10 Megabases, and no sequence should be larger than 2 Megabases.
Optional Fields
These options allow you to customize your VISTA analysis. You can use independently obtained gene annotations, select appropriate RepeatMasker options, give specific names to the analyzed sequences, and vary sequence conservation analysis parameters. If you do not fill in these additional options, we will use their default values.
Alignment Program
Depending on the specifics of your analysis (see details in "about" - link) you can choose one of the following alignment programs:
-
AVID -- global pair-wise alignment. If you choose to use this program, one of the sequences should be finished, all others can be either finished or in a draft format. For all finished sequences in the set, AVID generates all-against-all pair-wise alignments which can be displayed using any sequence as a base (reference). If some of the sequences are in a draft format, AVID will generated their alignment to the finished sequence which will be used as a base (reference). This is the only alignment program available through this server that can handle draft sequences.
LAGAN -- global pair-wise and multiple alignment of finished sequences. If some of the sequences are in a draft format your query will be redirected to AVID to obtain multiple - pairwise alignment. Multiple alignment will be visualized by VISTA that will calculate and display sequence conservation projected on any sequence you indicate as a base. This is the only alignment program available through this server that produces true multiple alignments.
Shuffle-LAGAN -- glocal alignment of finished sequences. It detects rearrangements and inversions in sequences while producing a global end-to end map. If you input several sequences all pair-wise combinations will be processed and results visualized in VISTA. This is the only alignment program available through this server that can detect rearrangements and inversions.
For each sequence you can select:
Name
Select names for your species that will be shown in the legend. We suggest that you use something meaningful, such as the name of an organism, the number of your experiment, or your database identifier. When you use a GenBank identifier to input your sequence, by default we will use it as a name of the sequence.
Annotation
If a gene annotation of the sequence is available you can submit it in a simple plain text format to be displayed on the plot
Each gene is defined by its start and end coordinates on the sequence, and the name listed on one line. A greater than (>) or less than (<) sign should be placed before this line to indicate plus or minus strand, although the numbering should be according to the plus strand. The exons are listed individually with the word "exon", after the start and end coordinates of each exon. UTRs are annotated the same way exons, with the word "utr" replacing "exon".
For example:
< 106481 116661 gene1 106481 106497 utr 107983 108069 exon 109884 110033 exon 111865 112023 exon > 39424 42368 gene2 39424 39820 exon 41401 42368 exon > 77817 81088 gene3 77817 78820 utr 79538 80107 exon
There is an easy way to export annotations in the above format from the Ensembl genome browser. Here is how it can be done:
- select your region of interest in the Ensembl browser;
- click on the "Export information about region" in the left part of the page;
- choose "VISTA Format" for the "Output Format";
- click the "Continue" button;
- click on the "Annotation data" link;
- save the result as a plain text file.
Our web server also accepts annotations in GFF3 format.
Repetitive elements (an option for RepeatMasker)
We recommend masking a base sequence to get better alignment results. You can submit either masked or unmasked sequences. If you submit a masked sequence and the repetitive elements are replaced by letters "N", select "one-celled/do not mask" option in the pull-down menu. We also accept softmasked sequences, where repetitive elements are shown as low-case letters while the rest of the sequence is shown in capital letters. In this case you need to select "softmasked" option in the menu.
If your sequences are unmasked, our server will mask repeats with RepeatMasker. Please select a specific mask for your base sequence in the menu. If you do not want your sequence to be masked, select "one-celled/do not mask".
Reverse-complement
Select the alignments for which you want to reverse-complement second sequence (try this if you get no homology when you expect to see it).
Regulatory VISTA (rVISTA) access.
Our server can predict transcription factor binding sites by running Regulatory VISTA (rVISTA) on the resulting alignments. There is a size limit for rVISTA of up to 20K. See rVISTA instructions for information regarding this tool.
-
-
Results
Several minutes after submitting your sequences you will receive email from vista@lbl.gov indicating your personal Web link to the location from where you can access results of your analysis.
Below is the results page. It lists every organism you submitted, and provides you with three viewing options using each organism as base. These three options are: Text Browser, which provides all the detailed information -- sequences, alignments, conserved sequence statistics, etc; VISTA Browser, which is an interactive visualization tool which can be used to dynamically browse the resulting alignments and adjust VISTA curve and conserved sequence parameters; and a PDF file, which is a static VISTA plot of the alignment.

At the bottom of the table there is a link that allows you to adjust conservation and visualization parameters. By clicking on it, the user can change certain parameters that are used to calculate conserved regions and to display VISTA graphs for each pair of submitted sequences. Please note that these parameters can also be adjusted on the fly when using VISTA Browser.
Text Browser
This link will bring you to the results of your analysis in the text format.

At the top of the page is a banner that displays the aligned organisms. The sequence listed in the darker header area is acting as base (to choose a different base, go back to the results page and click on the Text Browser link next to the desired base sequence name). This banner also lists the program used to align your sequences.

Underneath is the navigation area, which shows the coordinates of the currently displayed region, offers a link to the Vista Browser (see below), and a link to a list of all conserved regions found. In addition, if Shuffle-Lagan was used as the alignment program, there will be a link to download dot-plots of the produced alignments.

Following that is the main table, which lists each alignment that was generated for the base organism. Each row is a separate alignment. Each column, except the last one, refers to the sequences that were submitted for analysis. The last column contains information pertaining to the whole alignment.
The first cell of each row also contains a preview of the VISTA plot of this particular alignment, which allows one to quickly evaluate the quality of this alignment and to see alignment overlaps.
By looking at a row in this table, you can see which section of each organism aligned to which. The "Sequence" links will return a fasta-formatted piece of the organism sequence that participates in the alignment. Clicking on the "VISTA Browser" links will launch the VISTA browser set to show your curve with the selected organism as base, and the coordinates set to the coordinate of the selected alignment.
The last column provides links to alignments in human readable and MFA (multi-fasta alignment) formats, a list of conserved regions from this alignment alone, and links to pdf plots of this alignment alone. If the region being examined is 20K or less, rVISTA analysis can be performed, and a link to rVISTA will also be displayed here.
NEW! The last column also provides links to results of rankVISTA analyisis of the alignment. Read more about RankVISTA here.
VISTA Browser
Clicking on the VISTA Browser link will launch the applet with the corresponding organism selected as base. VISTA Browser is an interactive Java applet designed to visualize multiple alignments. The browser's clean display makes it easy to identify regions of high conservation across multiple species. Detailed help and instructions are available here: http://pipeline.lbl.gov/vgb2help.shtml
PDF
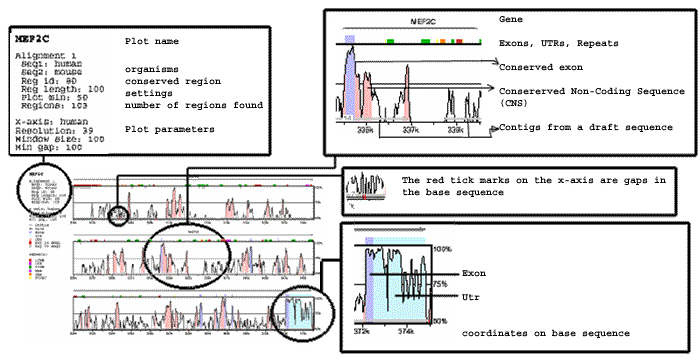
The PDF file is the visual representation of the alignment and conserved regions found. The most visible feature of the mVISTA picture is the "peaks and valleys" graph. This graph shows the percent of conservation (or percent difference, if you used the cVISTA option) between the two organisms at any given coordinate. The top and bottom percentage bounds are shown to the right of every row.
The coloring of the different conserved regions corresponds to the annotation of the region. By default, the pink regions are "Conserved Non-Coding Sequences" ("CNS"), the dark blue regions are exons, and the light-blue regions are UTRs. Gaps in the base sequence are signified by red sections of line underneath the plot. The color legend is summarized in the upper left-hand corner of the display
Arrows signifying genes are drawn above the graphs, pointing in the direction of the gene. Exons and UTRs are colored on them as well as on the main mVISTA plot. Gene names appear underneath the arrows if there is enough room. Repeats are shown directly above the plot, colored according the scheme to the left of the plot. The gray lines under the plot show contigs, which are numbered in the case of draft sequences.